Species Distribution Models
Source:vignettes/SpeciesDistributionModels.Rmd
SpeciesDistributionModels.RmdSpecies Distribution Modelling
Background
This vignette details the steps required to create a Species
Distribution Model (SDM) using the functions provided in
safeHavens. The SDM is required to use the
EnvironmentalBasedSample function, which is the most
complex sampling scheme provided in the package. The SDM is created
using an elastic net generalized linear model implemented via the
glmnet package. The SDM is then post-processed to create a
binary raster map of suitable and unsuitable habitat, that is used to
rescale the environmental predictor variables according to their
contributions to the model. These rescaled variables are then used in a
clustering algorithm to partition the species range into environmentally
distinct regions for germplasm sampling.
The goal of most SDM’s is to create a model that accurately predicts where a species will be located in environmental space and can then be predicted in geographic space. The goal of these models is to understand the degree and direction to which various environmental features correlate with a species observed range. This model is not intended to replace a well-structured and thought-out SDM; rather, it is intended to provide a quick model that can be used to inform sampling strategies.
About
While the authors are prone to creating many ‘advanced’ SDMs models
and in-house pipelines for developing them, we think that the juice is
not worth the squeeze and rely on a few powerhouse R packages to do the
heavy lifting. The main packages used are dismo,
caret, glmnet, CAST, and
terra. The dismo package provides a
user-friendly interface for working with species occurrence and raster
data, as well as some helper functions for thresholding and evaluating
models. The caret package provides a nice interface for
working with machine learning models, and the glmnet
package provides the elastic net model. The CAST package
provides functions for spatially informed cross-validation, which is
important for SDMs. The terra package provides functions
for working with raster data. All of these packages are well documented,
and maintained.
Steps to create an SDM
prep data
First you will need georeference occurrence data for your species of
interest. For common species we recommened the rgbif
package to download occurrence data from GBIF.
Here to maintain a smaller package size, and integrate with the
tutorial in dismo we use a built in data set for
Bradypus variegatus (Brown-throated sloth). We do this to also
leverage the raster data provided in dismo as well.
x <- read.csv(file.path(system.file(package="dismo"), 'ex', 'bradypus.csv'))
x <- x[,c('lon', 'lat')]
x <- sf::st_as_sf(x, coords = c('lon', 'lat'), crs = 4326)
planar_proj <- 3857 # Web Mercator for planar distance calcs
files <- list.files(
path = file.path(system.file(package="dismo"), 'ex'),
pattern = 'grd', full.names=TRUE )
predictors <- terra::rast(files) # import the independent variablesfit the model
Here we fit as SDM using elasticSDM. For arguments it
requires occurrence data x, a raster stack of
predictors, a quantile_v that is an offset
used to create pseudo-absence data, and a planar_proj used
to calculate distances between occurrence data and possible
pseudo-absence points. The quantile_v is used to create
pseudo-absence data by calculating the distance from each occurrence
point to the nearest neighbor, and then selecting a quantile of these
distances to create a buffer around each occurrence point. Points
outside of this buffer are used pseudo-absences.
sdModel <- elasticSDM(
x = x,
predictors = predictors,
quantile_v = 0.025,
planar_proj = planar_proj
)Under the hood this function uses caret to help out with
glmnet, as it provides output which is very easy to explore
and interact with. Elastic net modelS bridge the world of lasso and
ridge regression by blending the alpha parameter. Lasso has an alpha of
0, while Ridge has an alpha of 1. Lasso regression will perform
automated variable selection, and can drop (or ‘shrink’) features from a
model, whereas Ridge will keep all variables and if correlated features
exist split contributions between them. An elastic net blends this
propensity to drop or retain variables whenever it is used.
caret will test a range of alphas to determine how much
ridge or lasso regression characteristics are best for the data at
hand.
MAKE A NOTE ABOUT THE PCNM MORAN EIGNENVECTORS HERE TOO.
explore the output
{r Explore SDM output - Different alpha}sdModel$CVStructure coef(sdModel$Model, s = "lambda.min")
Here we can see how the elastic net decided that is the top model. We used Accuracy rather than Kappa as the main criterion for model selection.
sdModel$ConfusionMatrix
Confusion Matrix and Statistics
Reference
Prediction 0 1
0 18 2
1 5 20
Accuracy : 0.8444
95% CI : (0.7054, 0.9351)
No Information Rate : 0.5111
P-Value [Acc > NIR] : 3.102e-06
Kappa : 0.6897
Mcnemar's Test P-Value : 0.4497
Sensitivity : 0.9091
Specificity : 0.7826
Pos Pred Value : 0.8000
Neg Pred Value : 0.9000
Prevalence : 0.4889
Detection Rate : 0.4444
Detection Prevalence : 0.5556
Balanced Accuracy : 0.8458
'Positive' Class : 1
Here we can see how our selected model works on predicting the state
of test data. Note these accuracy results are slightly higher than those
from the CV folds. This is not a bug, CV folds are testing on their own
holdouts, while this is a brand new set of holdouts. The main reason the
confusion matrix results are likely to be higher is due to spatial
auto-correlation which are not address in a typical ‘random split’ of
test and train data. In order to minimize the effects of
spatial-autocorrelation on our model we use CAST under the
hood which allows for spatially informed cross validation.
Consider the output from CVStructure to be a bit more realistic.
binarize the output
terra::plot(sdModel$RasterPredictions)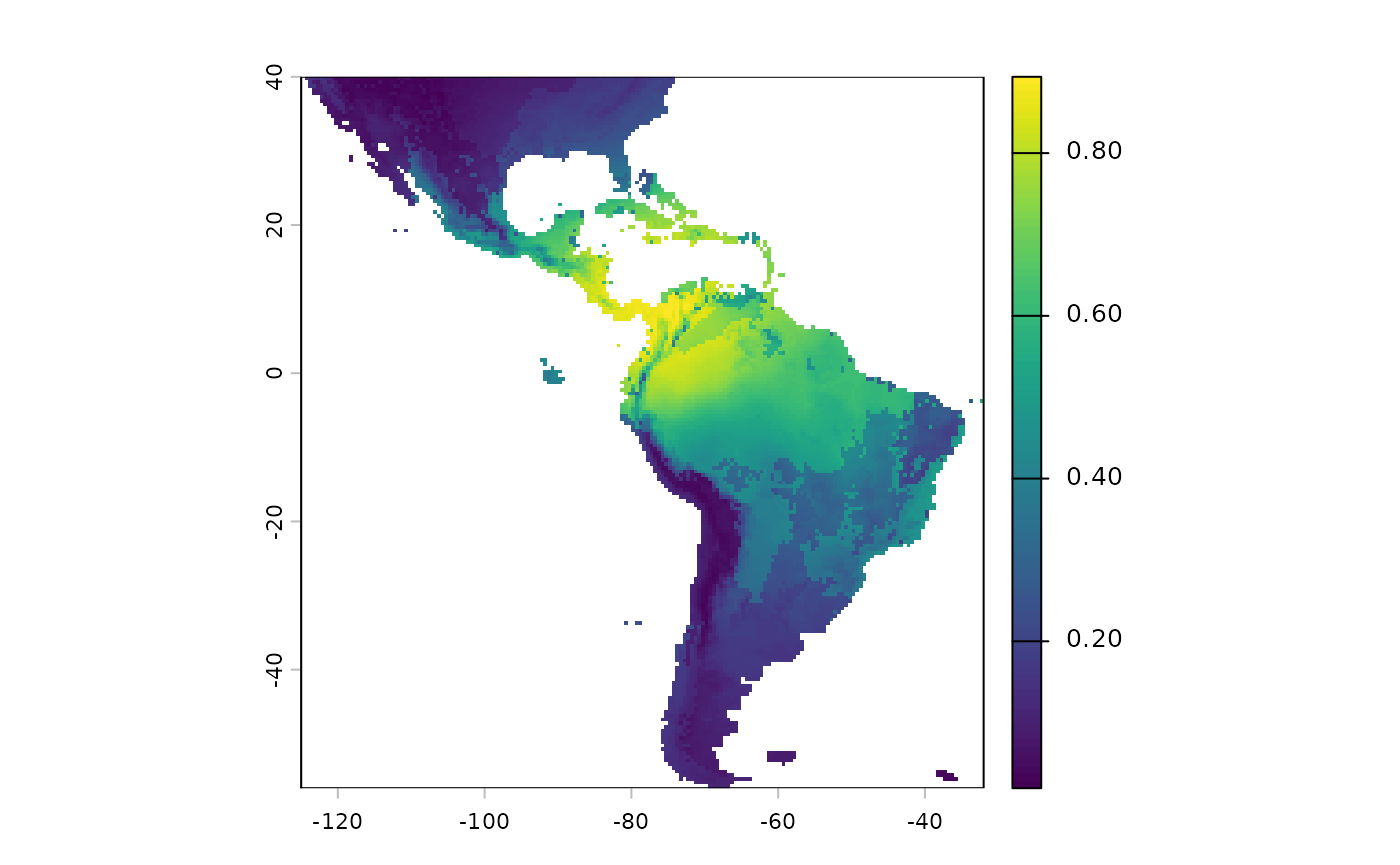
SDM’s produce continuous surfaces displaying the predicted
probability of suitable habitat across a landscape. However binary
surfaes (Yes/No Suitable), i.e. is it more or less probable that
suitable habitat for this taxon exists in this particular location (grid
cell)? are often required from them; the function
PostProcessSDM is used to binarize the predictions.
Going from a continuous surface to a binary surface loses information. The goal of the binary surface is to understand where the species is likely to be found, and then sample germplasm from these areas. There are many ways to threshold a SDM, and many caveats associated with this process; Frank Harrell has written on the topic of post-processing in statistics.
Historically, when assessing probability output 0.5 probability was
used as a threshold, probabilities beneath it considered ‘Not Suitable /
No’, while probabilities above are classified as ‘Suitable / Yes’. This
works for some cases, but thresholding is outside the domain of
statistics and in the realm of practice. My motto for implementing
models, is a slight elaboration of George Box’s aphorism, “All
models are wrong, some are useful - how do you want to be wrong?”.
Our goal of sampling for germplasm conservation is to maximize the
representation of allelic diversity across the range of a species. To
achieve this, we need an understanding of the species actual range,
hence it is better to predict the species present where it is not, than
to predict it absent where it actually grows. Accordingly, my default
thresholding statistic is Sensitivity. However, our function
supports all threshold options in dismo::threshold.
You may be wondering why this function is named
PostProcessSDM rather ThresholdSDM, the reason
for this discontinuity is that the function performs additional
processes after thresholding. Geographic buffers are created around the
intitial occurence points to ensure that none of the known occurrence
points are ‘missing’ from the output binary map. This is a two edged
sword, where we are again address the notion of dispersal limitation,
and realize that not all suitable habitat is occupied habitat.
What the PostProcessSDM function does is it again
creates cross validation folds, and selects all of our training data. In
each fold if then calculates the distance from each occurrence point to
it’s nearest neighbor. We then summarize these distances and can
understand the distribution of distances as a quantile. We use a
selected quantile, to then serve as a buffer. Area with predicted
suitable habitat outside of this buffer become ‘cut off’ (or masked)
from the binary raster map, and areas within buffer distance from known
occurrences which are currently masked are reassigned as probabilities.
The theory behind this process in underdeveloped and nascent, it does
come down to the gut of an analyst. With the Bradypus data set
I use 0.25 as the quantile, which is saying “Neighbors are generally
100km apart, I am happy with the risk of saying that 25km within each
occurrence is occupied suitable habitat”. Increasing this value
to say 1.0 will mean that no suitable habitat is removed, decreasing it
makes maps more conservative. The cost with increasing the distances
greatly is that the sampling methods may then puts grids in many areas
without populations to collect from.
threshold_rasts <- PostProcessSDM(
rast_cont = sdModel$RasterPredictions,
test = sdModel$TestData,
train = sdModel$TrainData,
planar_proj = planar_proj,
thresh_metric = 'sensitivity',
quant_amt = 0.5
)We can compare the results of applying this function side by side using the output from the function.
terra::plot(threshold_rasts$FinalRasters)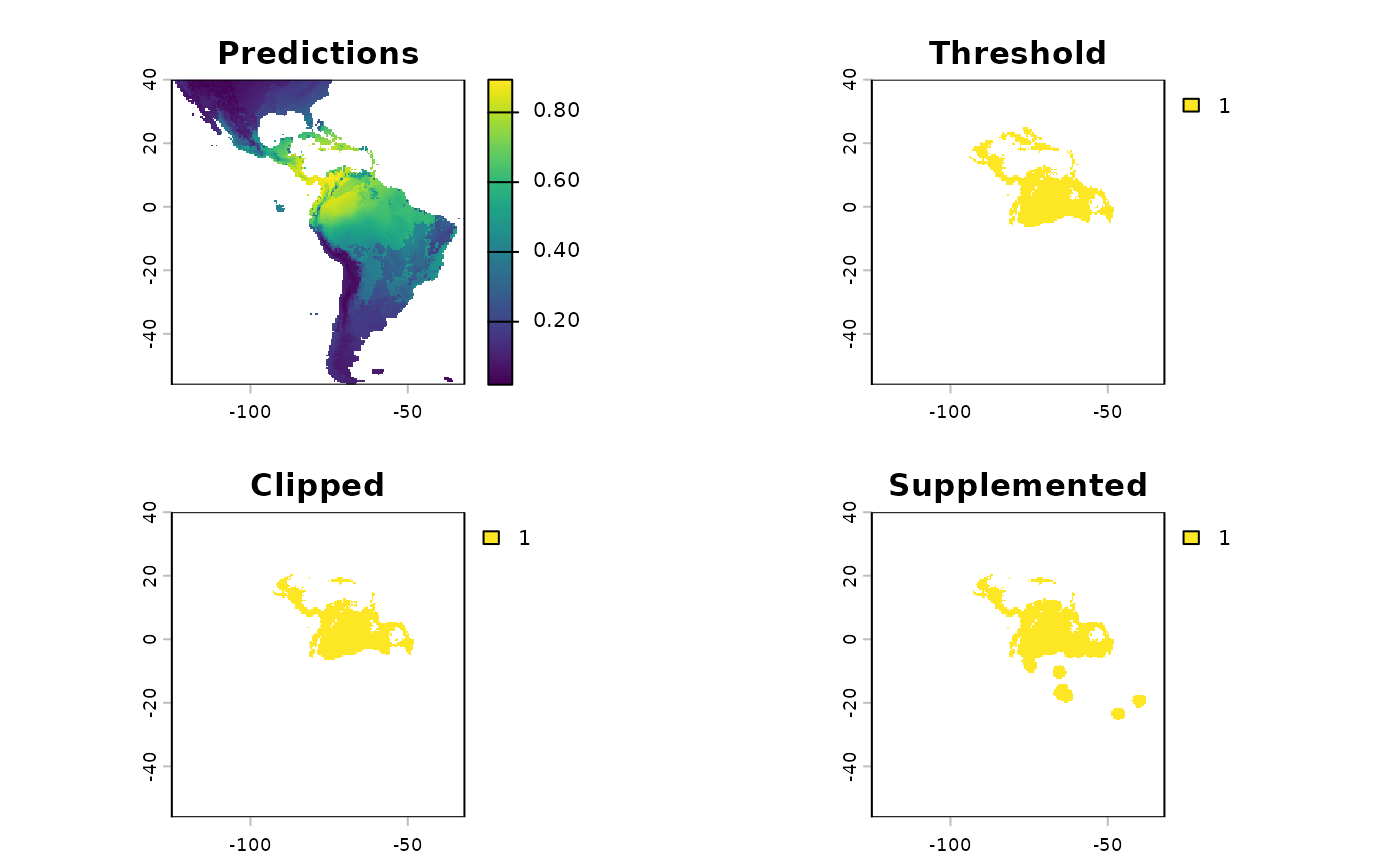
rescale predictor variables
glmnet is used for three main reasons, 1) it gives
directional coefficients and documents how each 1 unit increase in an
independent variable varies the response. 2) It maintains automated
feature selection reducing the work an analyst needs to do. 3)
glmnet re-scales all variables before model generation
combined with beta-coefficients allows for partitioning environmental
space into regions that are environmentally similar for the
individual species.
In this way our raster stack becomes representative of our model, we can then use these values as the basis for hierarchical cluster later on.
Below we create a copy of the raster predictions where we have
standardized each variable, so it is equivalent to the input to glmnet,
and then mulitplied by its beta-coefficient. We will also write out
these beta coefficients with the writeSDMresults function
right afterwards.
rr <- RescaleRasters(
model = sdModel$Model,
predictors = sdModel$Predictors,
training_data = sdModel$TrainData,
pred_mat = sdModel$PredictMatrix)
terra::plot(rr$RescaledPredictors)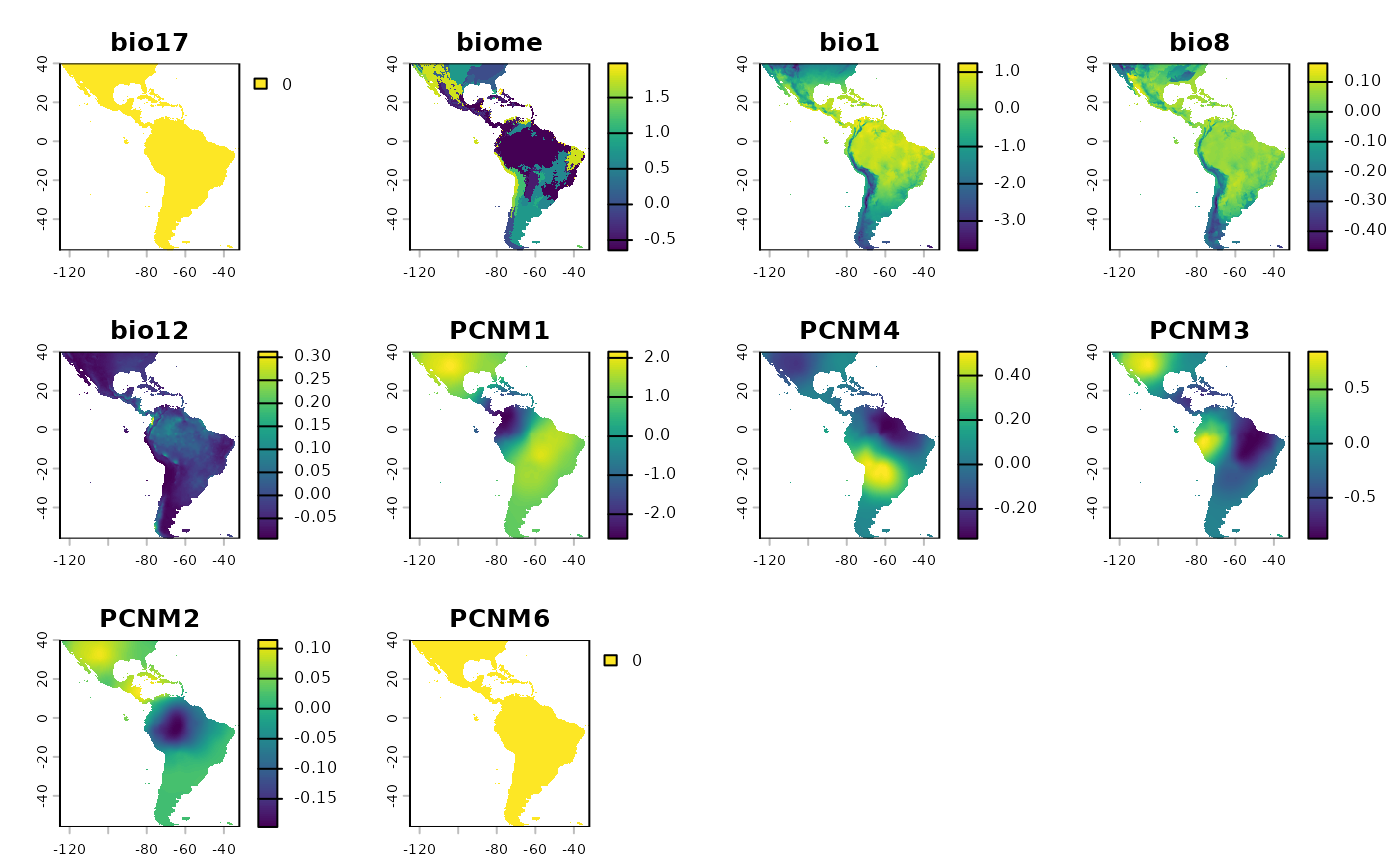
We can see that the variables are ‘close’ to being on the same scale, this will work in a clustering algorithm. If any of the layers are all the same color (maybe yellow?) that means they have no variance, that’s a term that was shrunk from the model. It will be dealt with internally in a future function. The scales of the variables are not exact, because they are weighed by their coefficients in the model
{r Show beta Coefficients from model print(rr$BetaCoefficients)
We can also look at the coefficients for each variable.
glmnet returns the ‘untransformed’ variables, i.e. the
coefficients on the same scale as the input rasters, we calculate the BC
right afterwards.
safeHavens generates all kinds of things as it runs
through the functions elasticSDM,
PostProcessSDM, and RescaleRasters. Given that
one of these sampling scheme may be followed for quite some time, it is
best practice to save many of these objects.
save results
bp <- '~/Documents/assoRted/StrategizingGermplasmCollections'
writeSDMresults(
cv_model = sdModel$CVStructure,
pcnm = sdModel$PCNM,
model = sdModel$Model,
cm = sdModel$ConfusionMatrix,
coef_tab = rr$BetaCoefficients,
f_rasts = threshold_rasts$FinalRasters,
thresh = threshold_rasts$Threshold,
file.path(bp, 'results', 'SDM'), 'Bradypus_test')
# we can see that the files were placed here using this.
list.files( file.path(bp, 'results', 'SDM'), recursive = TRUE )